-
Using the Diphosphanyl Radical as a Potential Spin Label: Effect of Motion on the EPR Spectrum of an R1(R2)P--PR1 Radical
L. Cataldo, C. Dutan, S.K. Misra, S. Loss, H. Grützmacher and M. Geoffroy
Chemistry - A European Journal, 11 (11) (2005), p3463-3468


DOI:10.1002/chem.200401276 | unige:3277 | Abstract | Article PDF
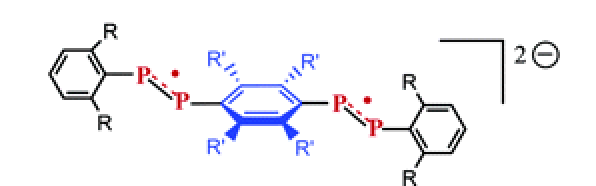
The EPR spectrum of the novel radical Mes*(CH3)PâPMes* (Mes*=2,4,6-(tBu)3C6H2) was measured in the temperature range 100-300 K, and was found to be drastically temperature dependent as a result of the large anisotropy of the 31P hyperfine tensors. Below 180 K, a spectrum of the liquid solution is accurately simulated by calculating the spectral modifications due to slow tumbling of the radical. To achieve this simulation, an algorithm was developed by extending the well-known nitroxide slow-motion simulation technique for the coupling of one electron spin to two nuclear spins. An additional dynamic process responsible for the observed line broadening was found to occur between 180 K and room temperature; this broadening is consistent with an exchange between two conformations. The differences between the isotropic 31P couplings associated with the two conformers are shown to be probably due to an internal rotation about the PâP bond.